How are DNA fragments separated in gel electrophoresis?
DNA electrophoresis is a standard laboratory technique used to identify, quantify, and purify DNA fragments. DNA electrophoresis involves loading DNA samples into the wells of an agarose or acrylamide gel and subjecting it to an electric field.
DNA fragments have a net negative charge; when subjected to an electric field, the negatively charged nucleic acid fragments migrate towards the positive electrode. The length of DNA fragments is a primary factor that influences the rate of migration—shorter DNA fragments travel faster through the gel matrix than longer fragments. Therefore, DNA electrophoresis results in separation—or resolution—of DNA fragments based on size.
We provides gels, reagents, and devices to support the DNA electrophoresis workflow.
precast gel technology allows rapid separation of DNA fragments and offers unparalleled convenience when compared to traditional DNA electrophoresis. Precast gels are available as Invitrogen E-Gel agarose gels, and Invitrogen TBE gel.
DNA loading buffers and tracking dyes
Loading buffers are solutions used for easy loading and tracking of DNA samples in agarose gels. Tracking dyes serve as visual markers of migration during electrophoresis and indicate when maximum resolution of the DNA fragments has been achieved. Additionally, visualization of DNA bands is not obscured by the tracking dyes because they run outside the limits of most DNA samples.
DNA Ladders for E-Gels
Invitrogen E-Gel DNA ladders are premixed with loading buffer and designed specifically for use with the E-Gel precast agarose gels in sizes ranging from 10 bp to 15 kb.
E-Gel DNA ladder selection guide
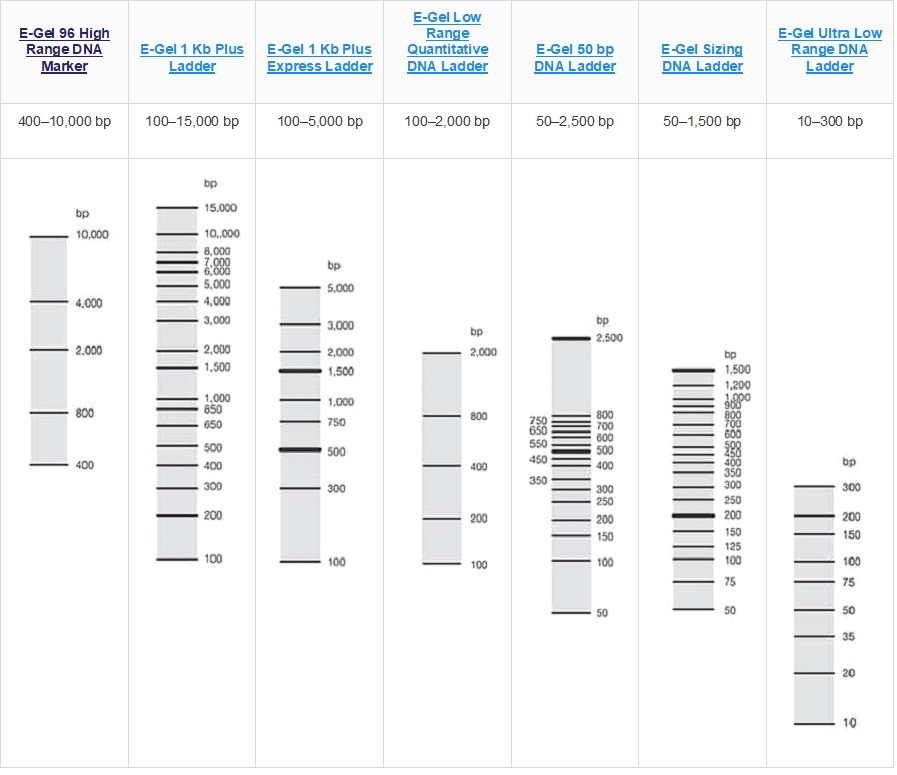
DNA ladder selection guide
The key to accurate band analysis is to use the correct DNA ladder suited for your particular application. This DNA ladder selection guide helps you choose the DNA ladder appropriate for your application.
| Product | Range, base pair (bp) | Ready to load (in loading buffer) | Tracking dyes |
|---|---|---|---|
| E-Gel DNA Ladders | 10 to 15,000 | Yes | Xylene Cyanol, Tartrazine (1X E-Gel Sample Loading Dye) or Xylene Cyanol, Orange G (1X E-Gel Sample Loading Dye) |
| TrackIt DNA Ladders | 1,000 to 10,000 (high mass) 100 to 2,000 (low mass) | Yes | TrackIt Cyan/Yellow Loading dye OR TrackIt Cyan/Orange Loading Dye |
| Mass DNA Ladders | 10 to 15,000 | No | Provided with 10X BlueJuice Gel Loading Buffer |
| DNA Ladders | 10 to 15,000 | No | Provided with 10X BlueJuice Gel Loading Buffer |
DNA & RNA Protocols
Find easy-to-follow protocols for everyday experiments including agarose protocols, DNA analysis protocols, DNA extraction protocols, mRNA protocols, and RNA protocols.
Agarose Protocols
- Agarose Gels
- Protein A Agarose
- See all
DNA Extraction Protocols
DNA Analysis Protocols
RNA Protocols
Introduction
| Product number | 610.11 | 610.12 |
|---|---|---|
| Dynabeads® Oligo (dT)25* | 5 ml | 10 ml |
| Lysis/Binding Buffer | 30 ml | 60 ml |
| Washing Buffer A | 60 ml | 120 ml |
| Washing Buffer B | 30 ml | 60 ml |
| 10 mM Tris-HCl (Elution Buffer) | 15 ml | 15 ml |
*Approximately 5 mg/ml, supplied in PBS pH 7.4, containing 0.02% NaN3 as a preservative.
Product # 610.11 provides enough reagents for 20 standard isolations.
Product # 610.12 provides enough reagents for 40 standard isolations.
The suspension of Dynabeads® Oligo (dT)25 and the buffers provided are produced and packed under RNase-free conditions. All kit reagents are of analytical grade and are RNase-free.
Intended Use
This product has been designed for a simple and rapid isolation of pure, intact polyadenylated (polyA) mRNA directly from the crude lysate of animal and plant cells and tissues. The isolated mRNA is suitable for use in all downstream applications.
Principle
The isolation protocol relies on base pairing between the polyA residues at the 3’ end of most mRNA, and the oligo (dT)25 residues covalently coupled to the surface of the Dynabeads® . Other RNA species lacking a polyA tail will not hybridize to the beads and are readily washed away. RNase inhibiting agents in the Lysis/Binding Buffer together with stringent hybridization and washing conditions ensure the isolation of pure, intact mRNA from crude samples rich in RNase, without the use of strong chaotropic agents. The protocol is flexible and can easily be scaled up or down to suit all sample sizes. It has successfully been used in the isolation of mRNA from single cells. The high capture efficiency facilitates detection of mRNA by reverse transcriptase (RT)-PCR from highly specialized cells (e.g. isolated from a heterogeneous sample by immunomagnetic separation). In addition, the protocol has been successfully used to isolate mRNA from a wide variety of tissues of mammalian, fish, amphibian, insect and plant origins.
For many applications elution of the mRNA from the beads is not required as the beads do not interfere with downstream enzymatic reactions. The bead-bound oligo (dT)25 can also function as a primer for RT and synthesis of first-strand cDNA, allowing the construction of solid-phase cDNA libraries and solid-phase RT-PCR.
Binding Capacity
1 mg of beads (200 μl) will bind up to 2 μg of mRNA. A typical mammalian cell contains about 10-30 pg of total RNA, from which 1-5 % is mRNA.
Description of Materials
Characteristics of Dynabeads® Oligo (dT)25
Dynabeads® are uniform, superparamagnetic beads. They are stable in the pH range of 4-13. Do not freeze the Dynabeads® Oligo (dT)25.
Diameter: 2.8 μm ± 0.2 μm (C.V. max 5%)
Surface area: 3-7 m2/g Density: Approx. 1.6 g/cm3
Magnetic mass susceptibility: 120 ± 25 × 10-6 m3/kg
Buffers
Lysis/Binding Buffer
100 mM Tris-HCl, pH 7.5
500 mM LiCl
10 mM EDTA, pH 8
1% LiDS
5 mM dithiothreitol (DTT)
Washing Buffer A
10 mM Tris-HCl, pH 7.5
0.15 M LiCl
1 mM EDTA
0.1% LiDS
Washing Buffer B
10 mM Tris-HCl, pH 7.5
0.15 M LiCl
1 mM EDTA
10 mM Tris-HCl (Elution Buffer)
10 mM Tris-HCl, pH 7.5
Please note that precipitate may form in the buffers. Dissolve precipitate before use by warming to room temperature and mixing thoroughly.
Additional Material Required
- Magnets: See www.lifetechnologies.com/magnets for magnet recommendations.
- RNase free pipette tips and pipettors.
- RNase free microtubes.
- Mixer allowing both tilting and rotation.
- Heat block and/or incubator at 65-80°C for elution step (if required).
For isolation of mRNA from tissue samples:
- Liquid nitrogen.
- Mechanical or manual tissue grinder.
- Syringe and 21 gauge needle.
- Benchtop microcentrifuge.
Protocols
Technical Advice
- Keep Dynabeads® Oligo (dT)25 in liquid suspension during storage and all handling steps. Resuspend well before use.
- Work RNase free and wear gloves.
- Bring all buffers, except the 10mM Tris-HCl (Elution Buffer), to room temperature prior to use. The 10mM Tris-HCl buffer should be stored on ice or at 2-8°C prior to use.
- Thorough resuspension of the beads/mRNA complex during washing and complete removal of the washing buffer at each step will prevent carry over of LiDS and other salts to the downstream reaction. Transferring the beads/mRNA complex to new tubes before the last washing step will further reduce LiDS carry over. LiDS is a strong inhibitor of enzymatic reactions.
Protocols included
Preparation of Sample Lysate
The mRNA content of cells and tissues varies greatly depending on the source of the material and RNA expression levels at the time of tissue/ cell harvest. Dynabeads® mRNA DIRECT™ Kit protocols can be scaled up or down to suit specific sample source and quantity. Please see section “Sample Guidelines and Scaling” before preparing the sample, and for recommended bead and buffer volumes (Table 1 and 2).
A) Preparation of Lysate from Solid Plant or Animal Tissue
- Aliquot (weigh) the animal or plant tissue while frozen, to avoid mRNA degradation. Ideally the tissue should be weighed and aliquoted before freezing. Do not exceed the specified amount of tissue, as using too much tissue will reduce the mRNA yield and purity.
- Grind frozen tissue in liquid nitrogen. Work quickly.
- Transfer the frozen powder to the appropriate volume of Lysis/Binding Buffer and homogenize until complete lysis is obtained (approx. 1-2 min). A rapid lysis in the Lysis/Binding Buffer is critical for preventing mRNA degradation.
- A DNA-shear step is advised for samples containing over 500,000 cells. Force the lysate 3-5 times through 21 gauge needle using a 1-2 ml syringe to shear the DNA. The reduction in viscosity should be noticeable. Repeated shearing causes foaming of the lysate due to detergent in the buffer, however, this should not effect the mRNA yield. The foam can be reduced by a 30 second centrifugation. The lysate can be frozen and stored at –80°C for later use.
- Prepare Dynabeads® Oligo (dT)25 as described below, and subsequently proceed with the mRNA isolation in “Direct mRNA Isolation Protocol”.
B) Preparation of Lysate from Cultured Cells or Cell Suspension
- Pellet cells by centrifugation (e.g. at 400 g for 8 minutes at 4°C) and wash the pellet by resuspending in phosphate-buffered saline (PBS). Pellet cells by centrifugation again. The cell pellet can be used immediately, or frozen in liquid nitrogen or at –80°C for later use.
- Add the appropriate volume of Lysis/Binding Buffer to either a frozen cell pellet or to a fresh cell pellet. Perform a repeated passage of the solution through a pipette tip to obtain complete lysis. The release of DNA during lysis results in a viscous solution which confirms complete lysis.
- A DNA-shear step is advised for samples containing over 500,000 cells. Force the lysate 3-5 times through a 21 gauge needle using a 1-2 ml syringe to shear the DNA. The reduction in viscosity should be noticeable. Repeated shearing causes foaming of the lysate due to detergent in the buffer, however, this should not effect the mRNA yield. The foam can be reduced by a 30 second centrifugation. The lysate can be frozen and stored at –80°C for later use.
- Prepare Dynabeads® Oligo (dT)25 as described below in section below, and subsequently proceed with the mRNA isolation.
Preparation of Dynabeads® Oligo (dT)25
- Resuspend Dynabeads® Oligo(dT)25 thoroughly before use.
- Transfer the desired volume of beads from the stock tube to a RNase-free 1.5 ml microcentrifuge tube and place the tube on a magnet (e.g. DynaMagTM-2).
- After 30 seconds (or when the suspension is clear), remove the supernatant.
- Remove the tube from the magnet and wash the beads by resuspending in an equivalent volume of fresh Lysis/Binding Buffer. Optional: For very small bead volumes (mini and micro isolations) use 50–100 μl wash volume to ease handling..
- Proceed to section below.
Direct mRNA Isolation Protocol
- Remove the Lysis/Binding Buffer from the pre-washed Dynabeads® Oligo(dT)25 by placing on the magnet for 30 seconds, or until the suspension is clear.
- Remove the microtube from the magnet and add the sample lysate
- Pipette to resuspend the beads completely in the sample lysate. Incubate with continuous mixing (rotating or roller mixer) for 3-5 min. at room temperature to allow the polyA tail of the mRNA to hybridize to the oligo(dT)25 on the beads. Increase the incubation time if the solution is viscous.
- Place the vial on the magnet for 2 min. and remove the supernatant. If the solution is noticeably viscous, increase the time to approx. 10 min.
- Wash the beads/mRNA complex two times with the appropriate volume of Washing Buffer A at room temperature. Use the magnet to separate the beads from the solution between each washing step.
- Wash the beads/mRNA complex once with the appropriate volume of Washing Buffer B at room temperature Use the magnet to separate the beads from the solution.
- If the isolated mRNA is to be used in enzymatic downstream applications (e.g. RT-PCR), one extra wash in Washing Buffer B is recommended. This should be followed by a final wash in the enzymatic buffer to be used (e.g. RT-PCR buffer without the enzyme or primers).
- If elution of mRNA from the beads is desired, add an appropriate volume of 10 mM Tris-HCl (Elution Buffer) and incubate at 65-80°C for 2 min. Immediately place the tube on the magnet, transfer the supernatant containing the mRNA to a new RNase free tube and place this tube on ice.
Re-use of Dynabeads® Oligo (dT)25 for Large Scale Isolations
Please note that the buffers supplied with the kit (product no. 610.11 and 610.12) may not be sufficient for large scale mRNA isolations. Multiple isolations from the same sample can be performed by re-using Dynabeads® Oligo(dT)25 after mRNA elution. Simply follow the protocol described in section “Direct mRNA Isolation Protocol”. After elution of the mRNA, wash the beads once in Lysis/Binding Buffer (section above). Add a new lysate sample to the beads and continue the isolation as usual. Alternatively, washed beads can be re-applied to the same sample lysate until all the mRNA has been captured.
Elimination of rRNA Contamination
In some cases trace amounts of ribosomal RNA have been observed in the mRNA samples. For many applications such as Northern blotting and RT-PCR, trace amounts of rRNA contamination will not interfere with the analysis or interpretation of the results. However, for other applications such as cDNA library construction and microarray analysis, rRNA contamination should be avoided. Ribosomal RNA is effectively eliminated by re-extracting the mRNA from the eluate. Re-use of the same Dynabeads® Oligo(dT)25 used for the original isolation is recommended. If new beads are used, it is recommended that the beads are washed in 50 mM Sodiumpyrophosphate before the isolation of mRNA.
- Follow the isolation protocol (Direct mRNA Isolation Protocol above). Elute the mRNA in 10 mM Tris-HCl (Elution Buffer). Transfer the eluted mRNA to a new tube and place on ice. Do not discard the beads.
- Wash the beads two times in Washing Buffer B.
- Dilute the eluted mRNA in 4 times its volume of Lysis/Binding Buffer (e.g. if the mRNA is eluted in 20 μl, add 80 μl of Lysis/Binding Buffer.)
- Remove the Washing Buffer B from the beads, by placing the tube on the magnet, and add the diluted mRNA Incubate with mixing at room temperature for 3-5 min.
Continue with the isolation protocol ( Direct mRNA Isolation Protocol, starting at step 4.)
Sample Guidelines and Scaling
The following information is intended as a rough guide to the expected total RNA content of selected tissues, as well as appropriate bead and buffer volumes.
Table 1. Estimated total RNA yield from mammalian cells and tissues
| Cell Types & Quantity | Estimated Total RNA Content (1-5% is mRNA) |
|---|---|
| Single mammalian cell | 10-30 pg |
| 50 mg of muscle tissue | 50-80 μg |
| 50 mg of liver tissue | 400 μg |
| 107 cultured fibroblasts | 50-80 μg |
| 107 cultured epithelial cells | 100-120 μg |
Table 2. Recommended volumes of Dynabeads® Oligo(dT)25 and buffers for use with different amounts of starting material
| Components | Maxi | Standard | Mini | Micro |
|---|---|---|---|---|
| Plant tissue | 100-400 mg | 20-100 mg | 4-20 mg | ≤ 4 mg |
| Animal tissue | 50-200 mg | 10-50 mg | 2-10 mg | ≤ 2 mg |
| Cultured cells | 4-20 × 106 | 1-4 × 106 | 0.15-1 × 106 | ≤ 150,000 |
| Dynabeads® Oligo(dT)25 | 1 ml | 250 μl | 50 μl | 10 μl |
| Lysis/Binding Buffer | 5 ml | 1250 μl | 300 μl | 300 μl |
| Washing Buffer A | 10 ml | 1-2 ml | 600 μl | 600 μl |
| Washing Buffer B | 5 ml | 1-1.5 ml | 300 μl | 300 μl |
| Tris-HCl (elution is optional) | 50-100 μl | 10-25 μl | 10 μl | 10 μl |
Sample Types from which mRNA has been Isolated Using Dynabeads® Oligo (dT)25
Table 3. mRNA DIRECT from animal tissues
| Tissue | Species | References |
|---|---|---|
| Adrenals | Rat | 4 |
| Brain | Mouse, Trout | 12, 4 |
| Brain (cereberal cortex, preoptic area, dentate gyrus) | Rat | 13, 14, 15 |
| Cartilage | Human | 16 |
| Organ of Corti and spiral ganglion | Guinea pig, rat | 17 |
| Ear (cochleae) | Mouse | 11 |
| Eggs | Trout | 4 |
| Gut (paraffin embedded) | Human | 18 |
| Heart | Rat | 13, 19* |
| Hypothalamus | Rat | 15 |
| Kidney | Rat | 13 |
| Kidney (glomerular preparations) | Human | 20 |
| Liver (paraffin embedded) | Human | 18 |
| Liver | Rat, trout, Xenopus | 13, 19*, 4 |
| Lung (paraffin embedded) | Human | 18 |
| Lung | Rat | 13 |
| Muscle | Rat, trout | 19*, 4 |
| Nematode (frozen rehydrated cysts) | Globodera rostochiensis | 58 |
| Ovaries | Trout, Xenopus | 4 |
| Pancreas | Rat | 19* |
| Paraffin embedded lung, liver, gut | Human | 18 |
| Paraffin embedded keratinocytes | Human | 21 |
| Pituitary | Rat | 22 |
| Plasma | Human | 59 |
| Pronephos | Trout | 4 |
| Skin (dried) | Frog | 57 |
| Spleen | Rat | 4, 13 |
| Trematode | Schistosoma mansoni | 23 |
| Whole insect | Drosophila | 4 |
*Lysis buffer with 4 M urea and 1 % SDS
Table 4. mRNA DIRECT from plant tissues
| Tissue | Species | References |
|---|---|---|
| Whole plants | Arabidopsis thaliana Rice, Oryza sativa | 23, 24, 25, 26 27 |
| Bud | Tobacco | 28 |
| Epidermal leaf cell (single cells) | Tomato | 1 |
| Embryos | Maize, tobacco | 29 |
| Flowers | Guinea pig, rat | 17 |
| Guard cell in leaf (single cells) | Tomato | 1 |
| Leaves | Barley Brassica oleracea Maize Potato Tobacco Tomato | 31, 4, 32 33 29, 27 34 28 1 |
| Ovules | Maize | 29 |
| Roots | Barley Brassica oleracea Spruce Maize | 31 33 6 29 |
| Seed aleurone | Barley | 31, 4, 35, 36, 32 |
| Seed endosperm | Barley | 35, 36, 32 |
| Seed embryos | Barley | 31, 4, 35, 36, 32 |
| Seedlings | Maize, tobacco | 29, 28 |
| Single leaf cells | Tomato | 1 |
| Stem | Tobacco | 28 |
| Stigma | Brassica oleracea | 33, 37 |
| Stolon tips | Potato | 34 |
Table 5. mRNA DIRECT from different types of cells
| Cell type/cell line | Origin | References |
|---|---|---|
| Chondrocytes | Human | 16 |
| Cervical cancer cells (HeLa) | Human | 38, 39 |
| Colon carcinoma cell line (COLO320) | Human | 40 |
| Fibroblast cells line (ST-1 and SKB-1) | Human | 41, 9 |
| Fibroblast (D551) | Human | 8, 9 |
| Fibroblast (RTG-2) | Trout | 4 |
| Endothelial cells (umbilical cord) | Human | 38, 9 |
| Hepatocyte cell line (HepG2) | Human | 8, 39 |
| Keratinocytes | Human | 42, 21 |
| Langerhans cells | Human | 42 |
| Lymphoblast B-cell lines (Reh, Daudi, HL-60, IM9) | Human | 8,4,39,43 |
| Mamma carcinoma cells (MCF7) | Human | 38, 39 |
| Mamma carcinoma (T47D) | Human | 40 |
| Monocytes | Human | 44 |
| Pancreas, insulinoma Rinm5F cells | Rat | 45 |
| Peripheral blood mononuclear cells (PBMC) | Human | 46 |
| Peritoneal exudate cells | Human | 42 |
| Placental cell line (AMA) | Human | 38, 39 |
| T-cells/T-cell clones | Human | 2, 47, 48, 49, 50 |
| Yeast (Saccharomyces cerevisiae, Hansenula polymorpha) | In soil samples | 51 |
| Yeast (Saccharomyces cerevisiae) | Culture | 52 |
Table 6. Direct isolation of viral polyA RNA with Dynabeads® Oligo (dT)25
| Starting Material | Virus | References |
|---|---|---|
| Cells in bronchoalveolar washes | HIV-1 | 47¹ |
| Cerebrospinal fluid | HIV-1 | 53¹ |
| Cell line | HTLV-I/II | 54² |
| Peripheral blood mononuclear cells (PBMC) | HIV-1 | 47¹ |
| Plasma | HIV-1/HIV-2 | 54¹ ² |
| Serum | HIV-1 | 55¹, 56¹, 54¹ ² |
| T-lymphocytes cell line (CD4+) | HIV-1 | 47¹ |
1) Lysis/binding buffer: 1 M LiCl, 2% SDS, 2xTE, 50 μg tRNA, Vanadyl ribonucleosyl complexes.
2) Lysis/binding buffer: 4 M GTC, 0.5% sarkosyl, 1% DTT, 0.5 M LiCl, 0.1 M Tris pH8.
Troubleshooting
| Problem | Possible Cause | Suggested Solution |
|---|---|---|
| Clumping of beads during incubation step with sample lysate. | DNA in the sample lysate has not been completely sheared. | i) Pipette the solution several times through a 1 ml pipette. ii) Increase force/number of passages through the needle in future shearing steps. |
| mRNA is contaminated with DNA. | i) Incomplete DNA shearing. ii) Incomplete removal of sample lysate after hybridization step, and subsequent carry over to wash and elution steps. iii) Inefficient washing. iv) Incomplete removal of wash buffers. v) Sample-to-beads ratio too high. | i) Increase the force and /or the number of passages through the needle in the DNA shearing step. ii) Completely remove the sample lysate after hybridization. iii) Make sure the beads/mRNA complex is fully resuspended in washing buffer. iv) Completely remove the sample/washing buffers. v) Dilute sample lysate or increase the amount of beads. vi) Re-extract the mRNA from the eluate. |
| mRNA yield is lower than expected. | i) Inefficient elution of mRNA from the beads. ii) Beads-to-sample ratio is too low. iii) Cells/tissue incompletely lysed. | i) Increase the elution volume/time/temperature or perform the elution step two times, pooling the eluate. ii) Increase the amount of beads. iii) Repeat the homogenization step. |
| The beads/cDNA complex is clumped and sticking to the tubes after reverse transcription. | Non-specific electrostatic interactions between the cDNA molecules and the plastic materials of the tubes/pipette tips. | i) Add BSA (0.2-1.0% final concentration) to the reverse transcription mix before performing the cDNA synthesis. This is to reduce clumping of the beads for a more efficient cDNA synthesis. Note: use best possible BSA quality. ii) Where appropriate, add 0.05% Tween-20 to the reaction buffers. iii) Alternatively, dilute the beads/cDNA solution (after reverse transcription) with an equal volume of the 1 × reverse transcription reaction buffer containing 0.05% Tween-20. Mix by pipetting and transfer the suspension to a new tube. If there are any remaining beads stuck to the tube walls, remove by washing with a fresh aliquot of buffer containing Tween-20. Pool these beads with the original bead suspension. Place the pooled beads on a magnet and remove the supernatant, then wash 2-3 times with the buffer containing Tween-20. Store the solid-phase cDNA library in an appropriate buffer containing 0.05% Tween-20. |
| Unable to detect specific mRNA molecules. | i) The beads-to-sample ratio is too low. ii) Inappropriate sample volume. iii) Hybridization time too short. | i) Increase the amount of beads. ii) Reduce sample volume/increase sample concentration. iii) Increase hybridization incubation time to 10-15 min. |
Comment
Using the appropriate bead-to-sample ratio (volume and concentration) there is no bias in hybridization based on mRNA size. However, with a large excess of mRNA or short incubation time, binding to the beads may be biased towards the short molecules. A similar bias may occur if the sample volumeto- bead ratio is too high.
General Information
Invitrogen Dynal® AS complies with the Quality System Standards ISO 9001:2000 and ISO 13485:2003.
Storage/Stability
This product is stable until the expiry date stated on the label when stored unopened at 2-8°C. Store opened vials at 2-8°C and avoid bacterial contamination. Keep Dynabeads® in liquid suspension during storage and all handling steps, as drying will result in reduced performance. Resuspend well before use.
Technical Support
Please contact Invitrogen Dynal® for further technical support (see contact details). Certificate of
Analysis/Compliance is available upon request.
Warning and Limitations
This product is for research use only. The product is not for use in human diagnostics or therapeutic procedures. Follow appropriate laboratory guidelines. This product contains 0.02% sodium azide (NaN3) as a preservative, which is cytotoxic. Avoid pipetting by mouth!
Sodium azide may react with lead and copper plumbing to form highly explosive metal azides. When disposing through plumbing drains, flush with large volumes of water to prevent azide build up. Material Safety Data Sheet (MSDS) is available at http://www.lifetechnologies.com.
References
- Karrer EE, et al. (1995) In situ isolation of mRNA from individual plant cells: Creation of cell-specific cDNA libraries. Proc Natl Acad Sci USA 92:3814-3818.
- Raineri I, et al. (1991) Improved efficiency for single-sided PCR by creating a reusable pool of first strand cDNA coupled to a solid phase. Nucleic Acids Research 19(14):4010.
- Lambert KN and Williamson VM. (1993) cDNA library construction from small amounts of RNA using paramagnetic beads and PCR. Nucleic Acids Research 21(3):775-776.
- Jakobsen KS, et al. (1994) Direct mRNA isolation using Magnetic Oligo(dT) Beads: A protocol for all types of cell cultures, animal and plant tissues. Advances in Biomagnetic Separation, Ed. Uhlèn M, Hornes E and Olsvik Ø, Eaton Publishing 61-71.
- Rodriguez IR and Chader GJ. (1992) A novel method for the isolation of tissue-specific genes. Nucleic Acids Research 20(13):3528.
- Sharma P, et al. (1993) PCR-based construction of subtractive cDNA library using magnetic beads. BioTechniques 15(4):610-611.
- Schraml P, et al. (1993) cDNA subtraction library construction using a magnet-assisted subtraction technique (MAST). Trends in Genetics 9(3):70-71.
- Aasheim HC, et al. (1994) A simple subtraction method for the isolation of cell-specific genes using magnetic monodisperse polymer particles. Biotechniques 16(4):716-721.
- Coche T, et al. (1994) Generation of an unlimited supply of a subtracted probe using magnetic beads and PCR. Nucleic Acids Research 22(7):1322-1323.
- Borgnes A, et al. Detection of isolated colon carcinoma cells in peripherial blood- or bone marrow mononuclear cell suspensions. Micrometastasis Meeting Munich, June 1996 (Abstract).
- Dazert SB, et al. (2007) Hearing development and spiral ganglion neurite growth in VASP deficient mice. Brain Research 1178:73-82.
- Moyal M, et al. (1992) Mutations in the UL53 gene of SV-1 abolish virus neurovirulence to mice by the intracerebral route of infection. Virus Research 26:99-112.
- Pines G, et al. (1992) Cloning and expression of a rat brain L- glutamate transporter. Nature 360:464-467.
- Alme MN, et al. (2007) Chronic Fluoxetine Treatment Induces Brain Region-Specific Upregulation of Genes Associated with BDNF-Induced Long-Term Potentiation. Neuronal Plasticity:1-9.
- Kim K, et al. (1994) Competitive PCR for quantification of gonadotropin releasing hormone mRNA level in a single micropunch of the rat preoptic area. Mol Cel Endocrinol 97:153-158.
- Winterpacht A, et al. (1994) Alter-native splicing as the result of a type II procollagen gene (COL2A1) mutation in a patient with Kneist Dysplasia. Hum Mol Genet 3(10):1891-1893.
- Safieddine S and Wenthold RJ. (1997) The glutamate receptor subunit d1 is highly expressed in hair cells of the auditory and vestibular systems. J Neurosci 17:7523-7531.
- de Andrès B, et al. (1995) Improved method for mRNA extraction from paraffin-embedded tissues. BioTechniques 18(1):42-44.
- Hengerer B. (1993) A rapid procedure for mRNA extraction from a large number of samples. Biotechniques 14(4):522-524.
- Bicknell G, et al. (1996) Separate extraction of mRNA from glomerular epithelial and mesangial cells. Pathological Society of Great Britain and Ireland, 172nd Meeting.
- Shamsher MK, et al. (1995) Novel mutations in keratin 16 gene underlie focal non-epidermolytic palmoplantar keratoderma (NEPPK) in two families. Hum Mol Genet 4(10):1875-1881.
- Schauder B, et al. (1994) Cloning of a cDNA encoding an ectoenzyme that degrades thyrotropin-releasing hormone. Proc Natl Acad Sci USA 91:9534-9538.
- Schüssler P, et al. (1995) Combined isolation of nucleic acids and protein from small amounts of tissue. Trends in Genetics 11(10):378-379.
- Rubin D. Magnetic Bead Isolation of mRNA from Plant Tissue. In: The Red Book Bulletin, Current Protocols in Molecular Biology, Subscriber’s Notebook,Anonymous 1992, p. 1,Suppl.20.
- Ivanov R. et al. (2008) EFFECTOR OF TRANSCRIPTION2 is involved in xylem differentiation and includes a functional DNA single strand cutting domain. Developmental Biology (313):93-106.
- Magrelli A, et al. (1994) Splicing of the rolA transcript of Agrobacterium rhizogenes in Arabidopsis. Science 266:1986-1988.
- Zhu QH, et al. (2007) compact shoot and leafy head 1, a mutation affects leaf initiation and developmental transition in rice (Oryza sativa L.). Plant Cell Reports (26):421-427.
- Hewelt A, et al. (1994) Promoter tagging with a promoterless ipt gene leads to cytokinin-induced phenotypic variability in transgenic tobacco plants: implications of gene dosage effects. Plant Journal 6(6):879- 891.
- Breton C, et al. (1995) PCR-generated cDNA library of transition-stage maize embryos: cloning and expression of calmodulin genes during early embryogenesis. Plant Mol.Biol. 27:105-113.
- Allen RL and Lonsdale DM. (1993) Molecular characterization of one31. Jakobsen KS, et al. (1990) Purification of mRNA directly from crude plant tissues in 15 minutes using oligo dT microspheres. Nucleic Acids Research 18(12):3669.
- Stacy RAP, et al. (1995) Evolution of the group 1 late embryogenesis abundant (Lea) genes: analysis of the Lea B19 gene family in Barley. Plant Mol Biol 28:1039-1054.
- Kumar V and Trick M. (1994) Expression of the S-locus receptor kinase multigene family in Brassica oleracea. Plant Journal 6(6):807-813.
- Bachem CWB, et al. (1994) Antisense expression of polyphenol oxidase genes inhibits enzymatic browning in potato tubers. Biotechnology 12:1101-1105.
- Espelund M, et al. (1992) Late embryogenesis-abundant genes encoding proteins with different numbers of hydrophilic repeats are regulated differentially by abscisic acid and osmotic stress. Plant Journal 2(2):241-252.
- Aalen R, et al. (1994) Transcripts encoding oleosin and a dormancy related protein are present in both the aleurone layer and the embryo of developing barley (Hordeum vulgare L.) seeds. Plant Journal 5(3):385-396.
- Giranton JL, et al. (1995) The S locus receptor kinase gene encodes a soluble glycoprotein corresponding t the SRK extracellular domain in Brassica oleracea. Plant Journal 8(6):827-834.
- Larsen F, et al. (1993) A tight cluster of five unrelated human genes on chromosome 16q22.1. Hum Mol Genet 2(10):1589-1595.
- Luna L, et al. (1994) Molecular cloning of a putative novel human bZIP transcription factor on chromosome m17q22. Genomics 22:553-562.
- Maas RA, et al. (1995) Immuno-magnetic purification of human breast carcinoma cells allows tumor-specific detection of multidrug resistance gene-1 mRNA by reverse transcriptase polymerase chain reaction in fine needle aspirates. Lab Invest 72(6):760-764.
- Slupphaug G, et al. (1991) Cell cycle regulation and in vitro hybrid arrest analysis of the major human uracil-DNA glycosylase. Nucleic Acids Research 19:5131-5137.
- Sprecher E and Becker Y. (1992) Detection of IL-1, TNF- and IL-6 gene transcription by the polymerase chain reaction in keratinocytes: Langerhans cells and peritoneal exudate cells during infection with herpes simplex virus-1. Arch Virology 126:253-269.
- Day S and Ross J. (1995) mRNA based method for typing the HLA-C locus gene by automated sequencing based typing. Eur J Immunogenet 23(1):92.
- Osnes LTN, et al. (1995) Lipopolysaccharide activation of human monocytes mediated by CD14, results in a coordinated synthesis of tissue factor TNF-alpha and IL-6. J Endotoxin Res 2:27-35.
- Bristulf J, et al. (1994) Interleukin-1 stimulates the expression of type I and type II interleukin-1 receptors in the rat insulinoma cell line Rinm5f; sequencing of a rat type II interleukin-1 receptor cDNA. Eur Cytokine Netw 5(3):319-330.
- Thanhäuser A, et al. (1993) Pentoxifylline: a potent inhibitor of IL-1 and IFNgamma biosynthesis and BCG-induced cytotoxicity. Immunology 80:151-156.
- Raineri I and Senn HP. (1992) HIV-1 promotor insertion revealed by selective detection of chimeric provirus-host gene transcripts. Nucleic Acids Research 20:6261-6266.
- Obata F, et al. (1993) A single universal primer for the T-cell receptor (TCR) variable genes enables enzymatic amplification and direct sequencing of TCR cDNA of various T-cell clones. Human Immunology 36:163-167.
- Spurkland A. (1992) Magnetic isolation of mRNA for in vitro amplification. Trends in Genetics 8(7):225-226. of the maize poly-galacturonase gene family members which are expressed during late pollen development. Plant Journal 3(2):261- 271.
- Murtagh JJ, et al. (1994) Alternative splicing of the guanine nucleotidebinding regulatory protein Goa generates four distinct mRNAs. Nucleic Acids Res. 22(5):842-849.
- Tebbe CC, et al. (1995) Direct detection of recombinant gene expression by two genetically engineered Yeasts in soil on the transcriptional and translational levels. Appl Environ Microbiology 61(12):4296-4303.
- Faulkner JDB and Minton NP. (1993) Rapid small-scale isolation of mRNA from whole yeast cells. Biotechniques 14(5):718-720.
- Chiodi F, et al. (1992) Human immuno-deficiency virus type 1 is present in the cerebrospinal fluid of a majority of infected individuals. J Clin Microbiol 30(7):1768-1771.
- Meijer A, et al. (1995) Development of molecular methods for detection and epidemiological investigation of HIV-1, HIV-2 and HTLVI/II infections. Report 118504 001 National Inst. Publ. Health and Env. Protection Bilthoven, The Netherlands.
- Albert JA, et al. (1992) Persistence of azidothymidine-resistant human immunodeficiency virus type 1 RNA genotypes in posttreatment sera. J Virol 66(9):5627-5630.
- Scarlatti G, et al. (1993) Comparison of variable region 3 sequences of human immunodeficiency virus type 1 from infected children with the RNA and DNA sequences of the virus population of their mothers. Proc Natl Acad Sci USA 90:1721-1725.
- Sin Y, et al. (2008) Skin bradykinin-related peptides (BRPs) and their biosynthetic precursors (kininogens): Comparisons between various taxa of Chinese and North American raid frogs. Peptides 29:393-403.
- Lu X, et al. (2007) A comparison of CFU-GM, BFU-E and endothelial progenitor cells using ex vivo expansion of selected cord blood CD133+ and CD34+ cells. Cytotherapy 9:292 – 300.
- Silva J, et al. (2007) Circulating Bmi-1 mRNA as a possible prognostic factor for advanced breast cancer patients. Breast Cancer Research 9:1-9.
- Jost R, et al. (2007) Magnetic quantitative reverse transcription PCR: A high-throughput method for mRNA extraction and quantitative reverse transcription PCR. BioTechniques 43:206-211.